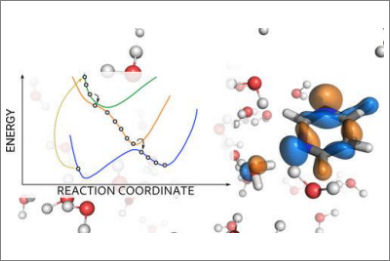
We present the implementation of trajectory surface-hopping nonadiabatic dynamics for a polarizable embedding QM/MM formulation. Time-dependent density functional theory was used at the quantum mechanical level of theory, whereas the molecular mechanics description involved the polarizable AMOEBA force field. This implementation has been obtained by integrating the surface-hopping program Newton-X NS with an interface between the Gaussian 16 and the Tinker suites of codes to calculate QM/AMOEBA energies and forces. The implementation has been tested on a photoinduced electron-driven proton-transfer reaction involving pyrimidine and a hydrogen-bonded water surrounded by a small cluster of water molecules and within a large water droplet.
Bondanza, M.; Demoulin, B.; Lipparini, F.; Barbatti, M. & Mennucci, B.
J. Phys. Chem. A 126,6780-6789 (2022) https://doi.org/10.1021/acs.jpca.2c04756
