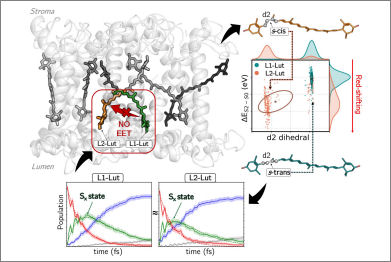
Carotenoid pigments are known to present a functional versatility when bound to light-harvesting complexes. This versatility originates from a strong correlation between a complex electronic structure and a flexible geometry that is easily tunable by the surrounding protein environment. Here, we investigated how the different L1 and L2 sites of the major trimeric light-harvesting complex (LHCII) of green plants tune the electronic structure of the two embedded luteins, and how this reflects on their ultrafast dynamics upon excitation. By combining molecular dynamics and quantum mechanics/molecular mechanics calculations, we found that the two luteins feature a different conformation around the second dihedral angle in the lumenal side. The s-cis preference of the lutein in site L2 allows for a more planar geometry of the π-conjugated backbone, which results in an increased degree of delocalization and a reduced excitation energy, explaining the experimentally observed red shift. Despite these remarkable differences, according to surface hopping simulations the two luteins present analogous ultrafast dynamics upon excitation: the bright S2 state quickly decays (in ~50 fs) to the dark intermediate Sx, eventually ending up in the S1 state. Furthermore, by employing two different theoretical approaches (i.e., Förster theory and an excitonic version of surface hopping), we investigated the experimentally debated energy transfer between the two luteins. With both approaches, no evident energy transfer was observed in the ultrafast timescale.
Pedraza-González, L.; Accomasso, D., Cupellini, L.; Granucci, G.; Mennucci, B.
Photochem Photobiol Sci (2023). https://doi.org/10.1007/s43630-023-00518-x
