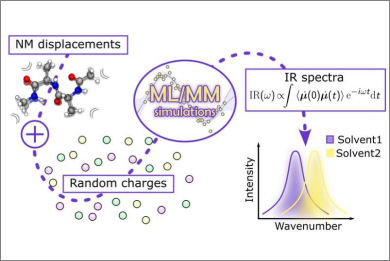
This work presents a multiscale machine-learning molecular dynamics (MD) strategy for the simulation of infrared (IR) spectra of solvated molecules. The approach combines efficient sampling of environmental configurations with a hierarchical model capable of predicting forces and dipole moments as analytical derivatives of the energy, enabling the direct simulation of IR spectra from MD trajectories.
Solvent effects are incorporated through a molecular mechanics (MM) representation of the environment embedded within the machine-learning (ML) description of the solute. This integrated ML/MM framework provides a balanced and computationally efficient treatment of solute–solvent interactions.
When applied to representative bio-related systems, the method reproduces experimental spectra with high fidelity and accurately captures solvent-induced vibrational shifts. The results demonstrate that this strategy offers a robust and cost-effective route for describing solvent effects in vibrational spectroscopy, paving the way for more accessible and reliable simulations of complex molecular systems.
Mazzeo, P.; Cupellini, L. & Mennucci, B.
Multiscale Machine Learning Prediction of Infrared Spectra of Solvated Molecules
Journal of Chemical Theory and Computation (2026)
Read the full paper here: https://doi.org/10.1021/acs.jctc.5c01959
