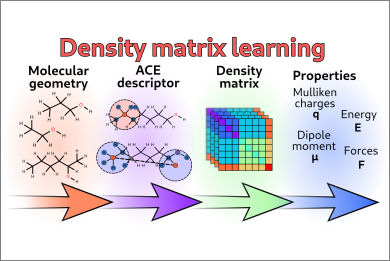
Calculation of energy, forces, and properties by Hartree-Fock and DFT methods requires solving the nonlinear self-consistent-field (SCF) equations, which is among the most computationally expensive steps. When many calculations on similar systems are required, machine-learning (ML) methods can be used to speed up these calculations; however, these methods require a suitable representation for the reduced density matrix, that is, the target of the SCF equations.
In this collaboration with the group of Prof. Bejamin Stamm (Univ. Stuttgart), we propose a symmetry-adapted representation of the density matrix, based on the atomic cluster expansion framework. Our representation not only preserves the basic symmetries of the density matrix elements, but can be also transferred to different molecules. We show in fact that we can use this representation with a simple linear regression model to learn the density matrix of previously unseen molecules. The predicted density matrix can be used as a guess to reduce the number of SCF iterations, or directly to calculate properties.
Zhang, L.; Mazzeo, P.; Nottoli, M.; Cignoni E.; Cupellini, L. & Stamm, B.
A symmetry-preserving and transferable representation for learning the Kohn–Sham density matrix
Digital Discovery (2026)
Read the full paper here: https://doi.org/10.1039/D5DD00230C
